Tutorial Module
Genomic Scoring Module
Before beginning the CarveAdornCurate (CAC) process, it is essential to complete the Genomic Annotation Scoring Module to ensure that reaction entries in the universal model are provided with reliable confidence scores. The CAC framework offers robust methodologies for constructing multiscale models that cater to both fungi and bacteria. A key feature of this approach is the necessity for distinct universal models and specialized genomic annotation tools tailored to the specific needs of the Genomic Scoring Module. To demonstrate this, we provide case studies featuring Corynebacterium glutamicum for prokaryotic models and Yarrowia lipolytica for eukaryotic models, showcasing the effective application of the genomic annotation scoring process.
C. glutamicum
Users must first select a bacterial genome file for analysis. In this example, we chose the genome file of C. glutamicum ATCC 13032 GCA_000196335.1_ASM19633v1_genomic.fna as the foundation for constructing the initial multiscale model. We then annotated this genomic file using two R scripts: generate_GSdraft.R and prepare_candidate_reaction_tables.R—provided by the gapseq tool, which is designed for building bacterial genome metabolic networks. The bitscore obtained from the annotation results plays a critical role as a scoring criterion for the reaction entries in the universal model. These annotation results are displayed in Figure 1. The genomic file, scripts, annotation results file gapseq_gene_annotation.csv. The universal model can either be directly used as the default one provided by gapseq, or it can be used in its modified form, such as the universal_model_icg that has been integrated with the classic genome-scale metabolic model of C. glutamicum>.
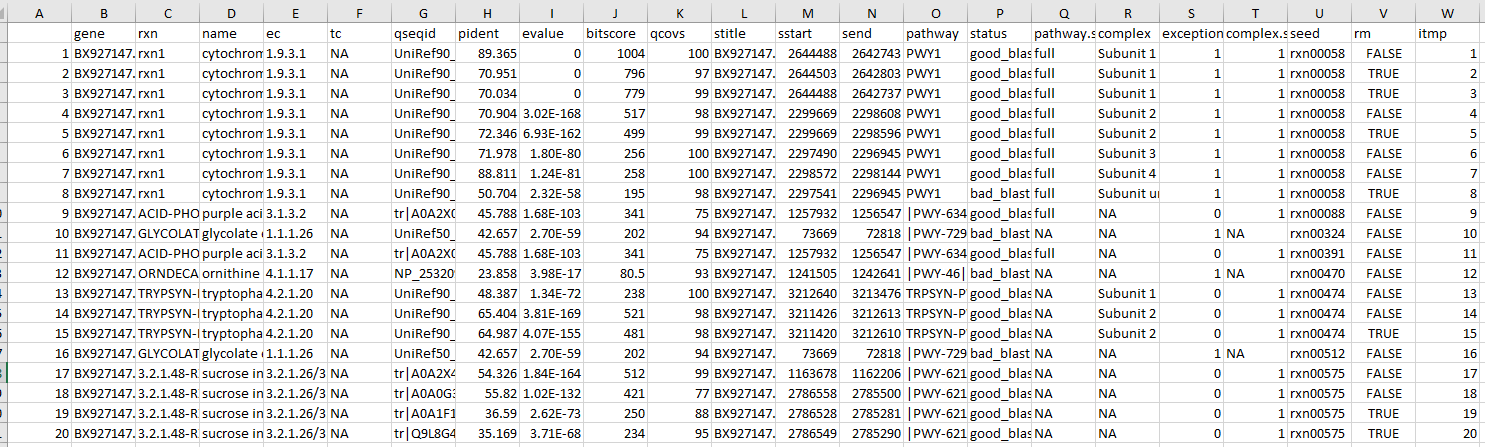
Figure 1. Genome annotation result of C. glutamicum
Y. ipolytica
In this study, we selected the Y. lipolytica DSM 3286 strain as the foundational basis for constructing a multiscale model. Owing to the absence of highly integrated tools such as gapseq for fungal model construction, we developed a bespoke scoring workflow using the sequence alignment software DIAMOND. Initially, we stored the sequences of the universal model in the file named new_fungi_seq.faa and subsequently created a sequence database, newfungiseq.dmnd. We then processed the Y. lipolytica genome file into a protein sequence file, formatted as Yarrowia_lipolytica_protein.faa. Using the DIAMOND command:
diamond blastx --db newfungiseq.dmnd --query Yarrowia_lipolytica_protein.faa --out out.tsv
The results of which are depicted in Figure 2. We further analyzed these alignment results using the Python script, Diamond_result_analysis.py, to generate the Yarrowia_lipolytica_model.xlsx. This file includes scores for the respective reactions within the unviersal model.

Figure 2. Genome annotation result of Y. ipolytica
Carve Module
The Carve module employs a Mixed Integer Linear Programming-based approach (MILP-gen) to generate ensembles of genome-scale metabolic models (GEMs) that are rich in genetic evidence, setting the stage for deeper insights. The Carve module aims to maximize the genomic score of the model while ensuring consistency with experimental data. This approach facilitates the construction of models that are both highly representative of the genomic data and aligned with empirical observations, thus enhancing the reliability and applicability of the generated models in biological research and application scenarios. Specifically, we integrated a manual-curated universal model that allows you to create high-quality model ensembles. This is done through a top-down approach where you can upload genome annotation files and cultivation conditions. Next, we will specifically introduce the functionalities of the Carve module by demonstrating the use of the data upload and download modules, respectively.
Upload data
The Carve module requires the upload of four types of data. The reaction scoring file, derived from the initial Genomic Scoring Module, must correspond to either a bacteria or fungal template. The culture conditions must specify the nutrients that the target organism can utilize, with data conforming to templates for bacteria or fungi. A universal model corresponding to either bacteria or fungi must be uploaded. Additionally, the size of the model ensemble needs to be chosen; more complex culture conditions necessitate more large matrix , which extends the computation time. Users aiming to fully leverage local computing power are advised to download and execute the relevant scripts from GitHub. The data upload interface is shown in Figure 3.

Figure 3. The data upload module of Carve.
Once your data is uploaded, the Carve module processes it on our server using the MILP gen method to construct a matrix and solve the model. Keep in mind, the solving time may vary depending on the size of your model set.
Download data
Once the solution process is complete, you can download the results right from the platform. The model set is stored in a file named "processed_file.csv". You can download the ensemble solution by clicking on the solution results shown in Figure 4.
Figure 4. The ensmeble solution download interface of the Carve module.
The solution result is presented in column format, where each column represents a distinct solution. This structure output makes it easy to analyze and apply the solutions to your research needs (Figure 5).

Figure 5. Display of the ensemble solution results.
Adorn Module
Next up, the Adorn module takes over to boost the ensemble’s performance by adding and fine-tuning various cellular processes and constraints. This step transforms basic GEMs into sophisticated multiscale models. The Carve and Adorn modules adopt CSV files for downloading and uploading model solution sets primarily because alternative formats, such as MAT files, tend to consume significantly more storage space and are less convenient for file transfer. Specifically, the Adorn module is primarily used to restructure classical GEMs and, on this basis, incorporate thermodynamic and enzymatic constraints to construct multiscale models.
Upload data
In the Adorn module, we have incorporated a multi-constraint integration framework that enables the integration of both thermodynamic and enzymatic constraints into the genome-scale metabolic models. The ensemble outputs generated by the Carve module can be directly utilized as inputs in the Adorn module. In addition, the Adorn module supports user customization of parameters involved in constructing enzyme-constrained and thermodynamically constrained models. Taking enzyme constraints as an example, enzyme turnover numbers (kcat) play a crucial role in the construction process. While kcat values can be experimentally measured, they are often derived from in vitro experiments and may vary across species for the same enzyme. Therefore, we recommend users consider utilizing advanced kcat prediction tools such as DLKcat to estimate these values. Of course, for users who prefer not to customize kcat data, Adorn also provides an automatic matching algorithm based on the GECKO framework. For thermodynamic constraints, Adorn allows users to define metabolite concentrations, which can be integrated with metabolomics data to build more accurate thermodynamic models. Users need not worry excessively about over-constraining the model, as Adorn offers a relaxation mechanism to ensure model feasibility. Alternatively, if users choose not to provide custom concentration values, Adorn can automatically assign reasonable concentration ranges to metabolites. All three types of data mentioned above can be uploaded directly via the Adorn module interface (see Figure 6).
Figure 6. The multiscale model ensemble download interface of the Adorn module.
Download data
Once the solution process is complete, you can download the results right from the platform. The ensemble of multiscale model is stored in a file named "processed_file.mat". You can download the ensemble solution by clicking on the solution results shown in Figure 7.

Figure 7. The data upload module of Adorn.
Curate Module
The Curate module employs machine learning methods to analyze simulation results from multiscale model ensembles with diverse network structures, thereby identifying model parameters with high curation value.
Upload data
The Curate module employs machine learning methods to analyze simulation results from multiscale model ensembles with diverse network structures, thereby identifying model parameters with high curation value.
Figure 8. The data upload module of Curate.
Download data
The output includes two columns of parameters: fractional importance and cluster ratio. A higher fractional importance indicates that the presence or absence of the parameter has a greater impact on the simulation results, and is therefore theoretically associated with higher curation value. A higher cluster ratio suggests that the parameter exhibits more complex interactions with other model parameters within the network. Users may determine the curation order based on the complexity of these interactions.

Figure 9. The data upload module of Curate.
Once the process is complete, you can download the results directly from the platform. The results include two parts: fractional importance and clustering coefficient. You can prioritize curating model parameters with high values for both.
We anticipate that CAC will become a digital cell construction platform capable of simulating complex cellular behaviors